
Theory and Modeling (TMG)
We use a variety of levels of theory, including electronic structure, molecular dynamics, electrodynamics and quantum dynamics, as well as machine learning and artificial intelligence approaches, to understand and predict nanoscale phenomena.
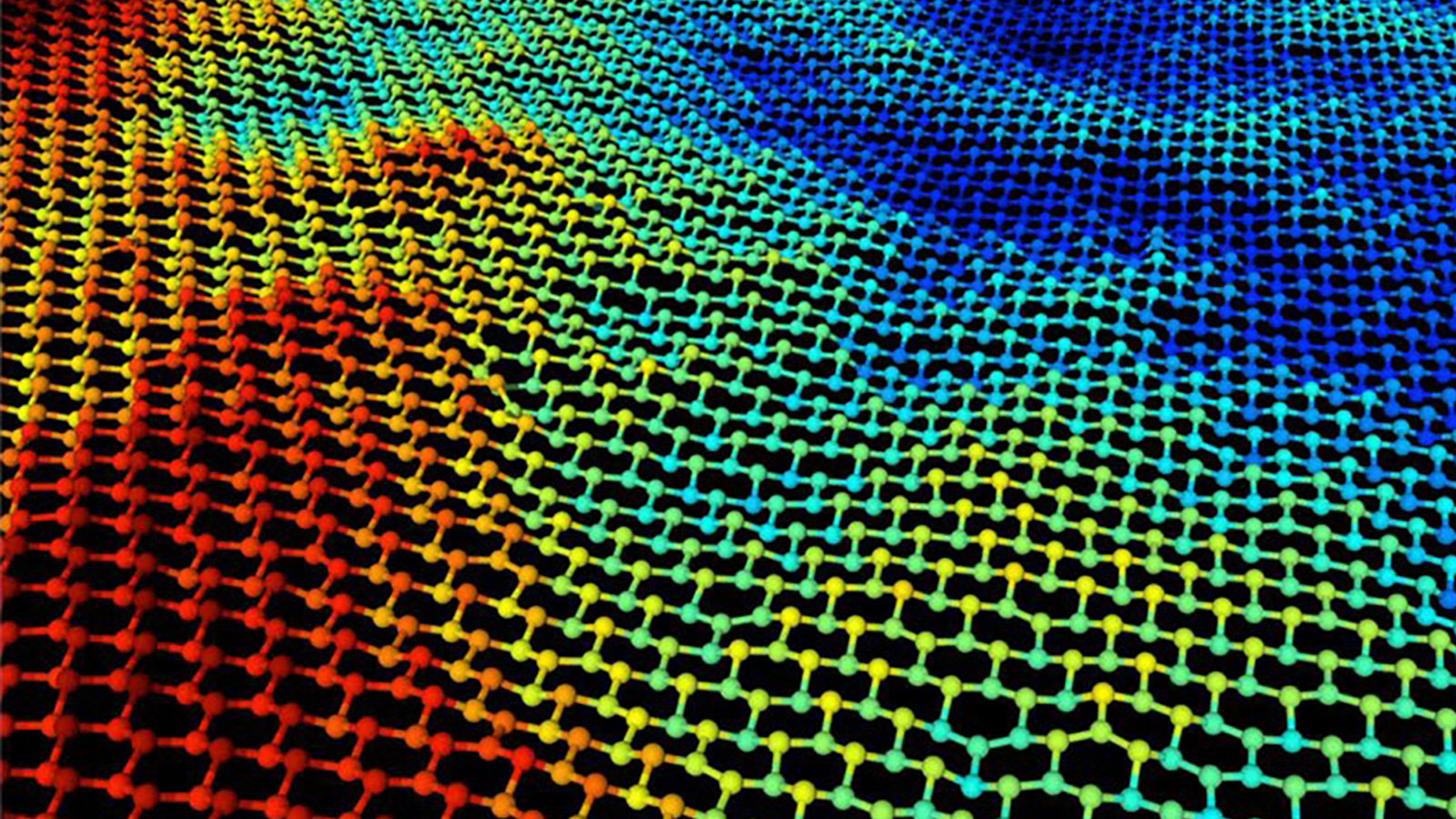
We apply and develop a broad range of state-of-the-art theoretical approaches for predicting the electronic structure, optoelectronic and dynamical properties of nanoscale materials. Additionally, we have expertise in machine learning or, more generally, artificial intelligence approaches for inverse design, force field development and the analysis of the large amounts of experimental data that arise from multimodal and ultrafast measurements.
Theory and modeling are essential for advancing nanoscience, not just in terms of providing explanations of experimental results, but also for leading to new experiments and directions. The methodology and capabilities that the Theory and Modeling group have, and are developing, are applied to scientific problems in the Center for Nanoscale Materials’ three themes that the group is particularly interested in. These include, but are not limited to:
- Quantum materials and sensing: probing entanglement dynamics, photon generation and mechanical cooling in hybrid quantum systems, quantum dot or, more generally, qubit interactions; using simulation to suggest new means for error mitigation in quantum systems.
- Manipulating nanoscale interactions: the control of mechanical energy dissipation at the nanoscale; learning how to manipulate light interactions in structured nanoscale arrays to achieve desirable outcomes such as flat lensing.
- Nanoscale dynamics: modeling and understanding of electron-induced heat for nanoscale thermal management and generation of non-equilibrium phase diagrams to guide the synthesis of metastable materials; incorporating first-principles data and experimental imaging data to yield precise atomistic-scale structures for real-time dynamics and other applications.
Key Capabilities
- Carbon, a High Performance Computing Cluster (30 teraflops)
- Electronic structure, molecular dynamics and electrodynamics codes
- Quantum dynamics and cavity quantum dynamics codes
- Machine learning based toolkits for force field development and atomistic imaging from multimodal data
Group capabilities
BLAST (Bridging Length/Timescales via Atomistic Simulation Toolkit)
Computational tool for bridging the electronic, atomistic and mesoscale simulations
CNM High Performance Computing Cluster (Carbon)
The CNM’s HPC Cluster "Carbon" offers capabilities for theory and modeling as well as for experimental data processing.
The cluster is located in CNM's dedicated data center in Building 440, occupying approx. 30% of the room's total area of 1,200 square feet. The cluster consists of several hundred individual computers and data storage units, placed in racks and tightly networked together to allow fast parallel processing. The hardware components are replaced or added over time to take advantage of more capable or more energy-efficient hardware.
Currently available for users are 2,600 cores on central processing units (CPU) and 20 Graphical Processing Units (GPUs), for approximately 25 Tflops performance, with 4–12 gigabytes of random access memory (RAM) per core and an InfiniBand interconnect for fast parallel computations and storage access. Storage is provided by a Lustre parallel file system with 520 terabytes net capacity, hosted on several Redundant Array of Independent Disks (RAID) systems and highly available servers.
A large number of HPC applications are installed on the cluster, with focus on atomistic modeling and nanophotonics. Packages for molecular visualization, general analysis, libraries, and development round out the system. Recent upgrades included addition of 40 nodes in FY 2017 and 52 nodes in FY 2018, and an upgrade of the operating system to CentOS-7.
Scientific Contact: Michael Sternberg
Dacapo
Dacapo is an ab-initio quantum mechanical molecular dynamics (MD) code using pseudopotentials and a plane wave basis set.
Density-functional-based tight-binding (DFTB)
Fast and efficient quantum mechanical simulation method (https://dftb.org/)
FANTASTX (AI/ML Framework framework to determine atomistic-level structures from multi-modal experimental and theoretical data)
Machine learning/artificial intelligence framework to determine atomic structures from experimental (currently: XAS, XRD, PDF, STEM, STM) data and simulations
GPaw, a real space, grid-based DFT-PAW code
GPAW is based on the projector-augmented wave method and can solve the self-consistent density functional theory (DFT) equations using three different wave-function representations, namely real-space grids, plane waves, and numerical atomic orbitals.
Ingrained
An open-source automation framework which solves for this correspondence and fuses atomic resolution image simulations into the experimental images to which they correspond
Time-domain nanophotonics simulation package
Tool for modeling nano-scale optical devices that solves Maxwell's equations directly.
VASP, ab-initio molecular dynamics calculations
Electronic structure simulation tool for performing ab initio quantum mechanical calculations using either Vanderbilt pseudopotentials, or the projector augmented wave method, and a plane wave basis set